Cell division by mitosis produces daughter genetic clones, using gatekeeper genes via that control the cell cycle via checkpoints. Mutations in DNA can disrupt these controls, resulting in uncontrolled cell growth, malignant tumours and cancer.
Each cell in the body divides a finite number of times (Hayflick limit). During each cell cycle a complex series of events takes place inside the cell, which ensures that each daughter cell is an exact genetic replica of the parent.
These events are orchestrated by master genes, which produce proteins that act as gatekeepers at the checkpoints between the different phases of the cell cycle.
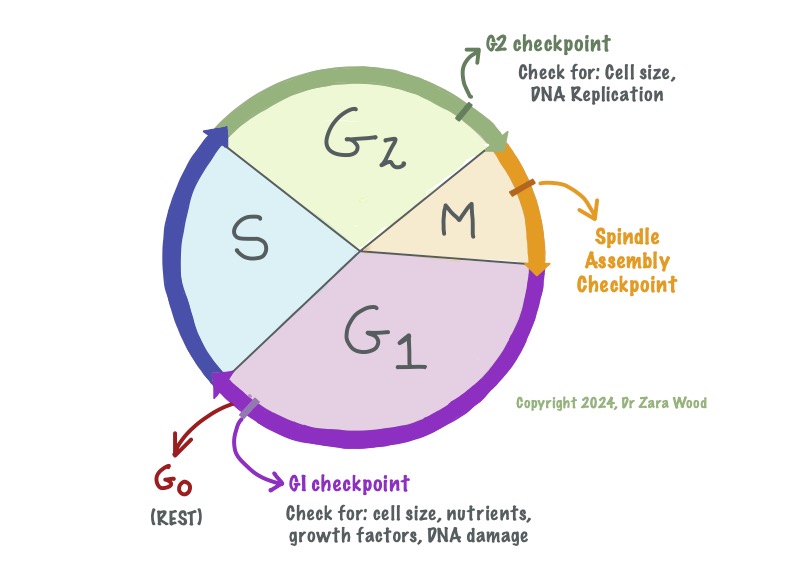
G1 and G2 are the growth phases, where new proteins and organelles are synthesised. S is the phase for DNA replication. M is Mitosis, where the DNA is divided between the daughter cells, so form two identical clones. G0 is the rest phase – cells are alive, but not actively dividing. When needed (ie a cut to the skin), cells can leave G0 and re-enter the cell cycle to produce new daughter cells.
How the gatekeeper genes work –
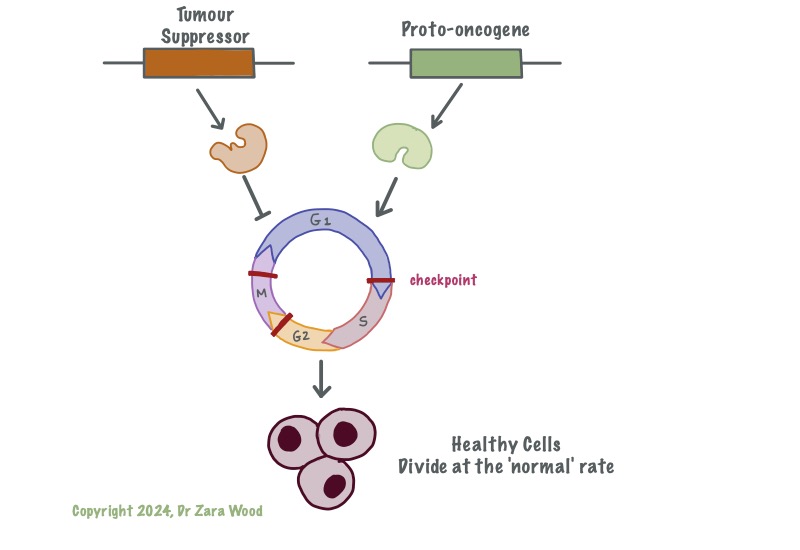
Tumour-suppressor genes act as down-regulators of the cell cycle, ensuring repairs are made before the cell proceeds to the next stage of the cell cycle. Tumour-suppressor genes are permanently switched on, and each cell has two functional copies of these genes. Proto-oncognes act as up-regulators of the cell cycle, ensuring it does not stall. Proto-oncogenes are turned on at critical checkpoints, this is usually in response to either internal or external signal proteins.
Functional tumour suppressor genes are critical in ensuring that the cell cycle runs smoothly. If an error is made during one of the phases in the cell cycle, the tumour suppressor genes apply the brakes to the cell cycle, and do not allow the cycle to continue unless the damage has been repaired. In case the damage is non-reversible, the tumour suppressor genes trigger the pre-programmed suicide cycle – called apoptosis – within the cell.
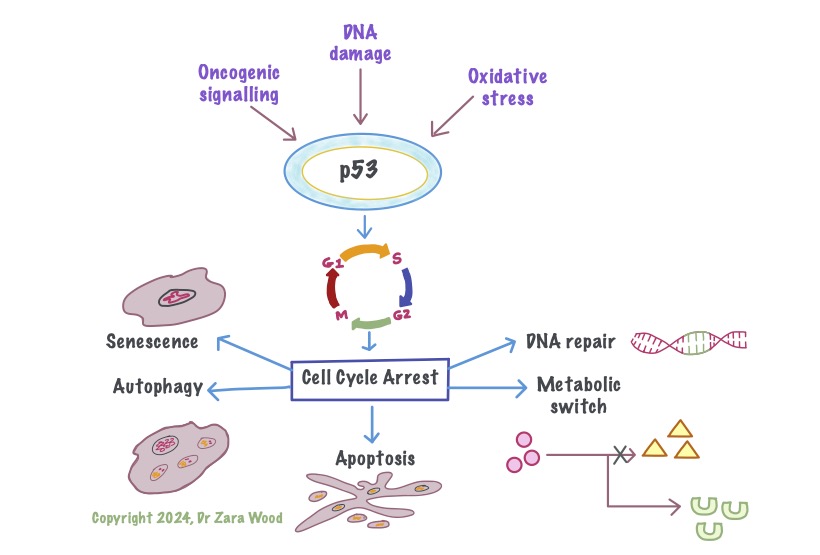
p53 is a tumour-suppressor gene, that down-regulates the cell cycle. For example, damage to DNA can trigger cell cycle arrest between the G1 and S phase. This gives time for the repair enzymes to repair DNA. If the DNA cannot be repaired, the apoptosis pathway is activated, which is a form of programmed cell death. Apotosis causes minimal damage to surrounding cells. Metabolic stress, like exposure to free radicals, or aging, can trigger autophagy or senescence in cells. They are generally considered tumour-suppressive, but in some cases can lead to chronic inflammation which damages surrounding tissue.
This ensures that damaged cells with defective DNA or protein are not able to propagate within the body.
By default, tumour suppressor genes are switched ‘ON’ all the time. Each one of us has two copies of this gene, one on each chromosome. A mutation may cause one of the copies to become non-functional, but this may not necessarily lead to cancer as the second copy of the gene is still functional. A mutation in the second copy, however, does not bode well for the cell. It effectively removes any brakes on the cell cycle, allowing damaged cells to continue to divide before the damage is repaired.
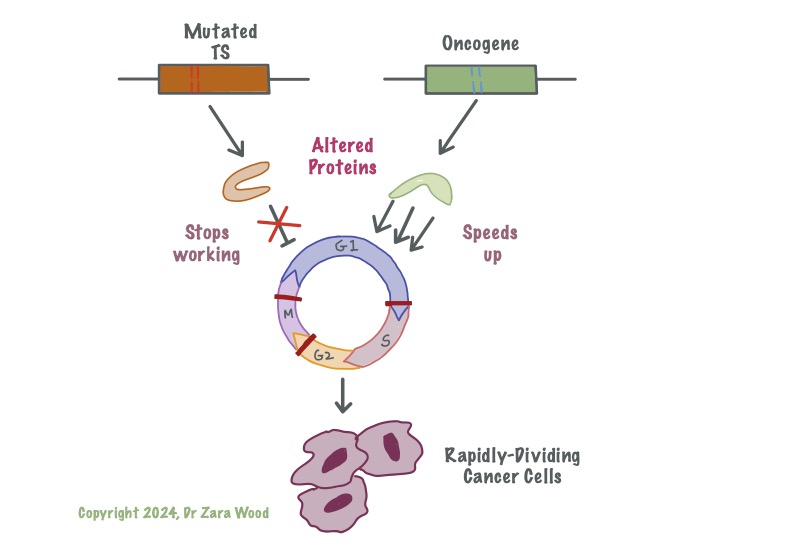
Mutation in the Tumour-suppressor genes means these genes no longer produce functional protein. Damage to one of the genes increases the risk of cancer, but leaves cells with one functional copy. Damage to both copies disrupts the cell’s ability to slow down the cell cycle – i.e. mutation in both copies of Tp53 gene means cells are no longer able to respond to DNA damage or oxidative stress. This allows cells with faulty DNA to continue to multiply. Proto-oncogenes are turned on at critical junctions, in response to internal or external signal proteins. A photo-oncogene with a mutation is called an oncogene. This oncogene might produce protein that is no longer under the control of the signal protein, and remains activated at all times. This can lead to uncontrolled cell division. Cells with mutation in either gene are able to outcompete healthy body cells for oxygen and nutrients, suffocating the healthy cells and causing cell death – this is cancer.
Over time, the proportion of damaged cells in relation to healthy cells increases. These defective cells may then start to grow rapidly and out-compete neighbouring ‘healthy’ cells for oxygen, glucose and other nutrients. This mass of cells is called a tumour. A tumour is benign if the cell growth is self-limiting. However, in some cases, the cells continue to grow indefinitely, and at a faster rate than normal cells – this is a malignant tumour, or cancer. If the malignant cells enter the lymphatic system and spread around the body, the cancer is said to have metastasised.
Common examples of tumour suppressor genes include TP53 and BRCA-1 and BRCA-2. Over half of human cancers are linked to defective p53 protein produced by the TP53 gene. BRCA-1 and BRCA-2 are linked to early-onset breast cancer and ovarian cancer. In very rare instances, children may be born with defective p53 genes, greatly increasing their susceptibility to cancer – this is called the Li-Fraumeni-Syndrome.
FURTHER READING:
- Chial, H. (2008) Tumor suppressor (TS) genes and the two-hit hypothesis. Nature Education 1 (1), 177
- Jing, K., and Lim, K. (2012) Why is autophagy important in human diseases? Exp Mol Med 44, 69–72
- Carlos, L et al. (2013) The Hallmarks of Aging. Cell, 153 (6), 1194 – 1217
- Aubrey, B. et al. (2018) How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ 25, 104–113
- Gasek, N.S. et al. (2021) Strategies for targeting senescent cells in human disease. Nat Aging 1, 870–879
- National Institute on Aging (2021) Does cellular senescence hold secrets for healthier aging?
- Panatta, E., et al. (2021) Understanding p53 tumour suppressor network. Biol Direct 16, 14
- Cell Signaling Technology (accessed 2024) Cellular Senescence
